



Исследование самоорганизации нанотубулярных аккумуляторов водорода с помощью модуля «Молекулярная наномеханика»
Содержание
I. Обоснование и проблематика
Большинство теоретических работ не привносят практически никакой ясности в вопрос о механизме взаимодействия водорода и УНТ. Природа связей между водородом и углеродом, а также внутри водородного адсорбата является отдельным спорным вопросом. Физическая и химическая сорбции предлагаются большей частью исследователей как два возможных механизма для процессов, лежащих в основе накопления водорода нанотубулярным углеродом. Однако, базируясь на данных классических представлениях, невозможно объяснить случаи достижения водородной емкости свыше 8 масс.%, т.е. образование суперадсорбата или значение энергий связи между водородом в углеродном нанотубулене, соответствующих «супер» физическому или слабому химическому взаимодействию [1, 2]. Для снятия существующих разногласий между результатами практического и теоретического экспериментов, необходимы новые подходы для объяснения взаимодействий водорода и УНТ.
Целью работы является выявление возможности существования углеродных нанотубулярных аккумуляторов бирадикалов водорода со значениями энергий связи в системе соответствующих экспериментальным значениям «супер» физического или слабого химического взаимодействия. В качестве одной из задач выступает проверка применимости модели стохастической диссипативной эволюции наносистем к описанию структурной организации исследуемой системы в различных температурных режимах.
II. Порядок выполнения работы
-
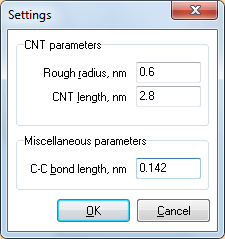
Рис. 1. Вид окна SettingsЗапустите программу CNTH. Первым шагом является задание параметров структуры нанотрубки — радиуса, длины, а также межатомного расстояния C—C, если оно должно отличаться от значения по умолчанию (0.142 нм).
Для указания этих параметров вызовите команду Settings… из меню Edit. В появившемся окне нужно ввести значение радиуса, длину нанотрубки и при необходимости — межатомное расстояние C—C.
-
Задав параметры, можно сконструировать готовую модель нанотрубки, вызвав команду Create CNT. CNTH позволяет при необходимости отключать отображение межатомных C—C-связей, оставляя лишь отображение ядер атомов углерода в виде точек. Данная возможность доступна при использовании команды Display bonds из меню View.
-
Рис. 2. Фрагмент структурыСуть процедуры конструирования модели адсорбата сводится к ручному заданию положений H-атомов бирадикала для одного лишь произвольного гексагона нанотрубки (C6) и последующему автоматическому распространению созданного таким образом правила на все остальные гексагоны. Пояснить порядок выполнения процедуры задания относительных координат лучше всего на конкретном примере. Требуется построить модель нанотубулярного аккумулятора, в котором бирадикалы располагаются непосредственно над C-атомами с внутренней стороны трубки, таким образом, что один на шестиугольник приходится три бирадикала.
-
Заметим, что каждый бирадикал в такой структуре «принадлежит» одновременно трем гексагонам, стыкующимся в точке расположения углеродного атома под водородом. Следовательно, на один гексагон приходится 6/3 = 2 атома H. Это в свою очередь означает, что нужно задать координаты только двух H-атомов для произвольно выбранного гексагона.
-
Вызовите команду Configure adsorbate… из меню Edit.
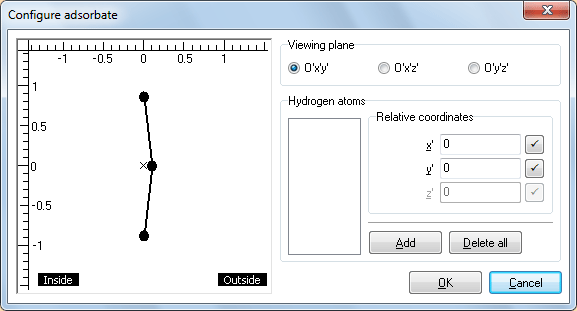
Рис. 3. Диалоговое окно Configure adsorbate -
В левой части появившегося окна Configure adsorbate находится изображение проекции гексагона C6 ранее построенной нанотубки. Плоскость проецирования можно переключать, выбирая нужную из группы Viewing plane. Вспомогательные надписи Inside и Outside на изображении поясняют, какая сторона гексагона обращена внутрь нанотрубки, а какая наружу. Крестиком × обозначено расположение начала относительных координат O'. Сами относительные координаты для наглядности отмеряются линейками, расположенными слева и сверху.
Группа Hydrogen atoms позволяет добавлять атомы водорода и управлять их относительными координатами x', y', z'. Добавьте два H-атома, последовательным двукратным нажатием на кнопку Add. Оба атома появятся в списке в левой части группы Hydrogen atoms, а также будут отрисованы в виде кружков на схеме проекции гексагона. Далее двукратным нажатием по любому из атомов в разделе Hydrogen atoms переводим Н в Н1 (Это необходимо для моделирования бирадикала Н↑—Н↓). Атомы на схеме можно перетаскивать указателем мыши.
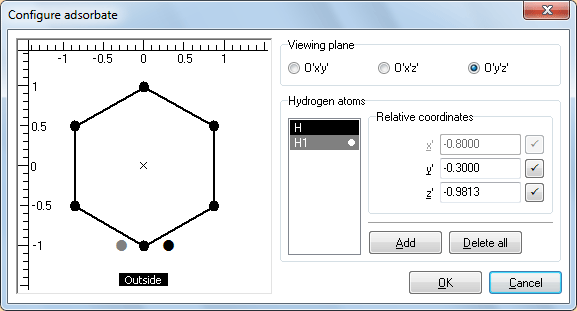
Рис. 4. Диалоговое окно Configure adsorbate -
Когда положения атомов водорода будут правильно отображаться на всех трех проекциях, нажмите кнопку OK.
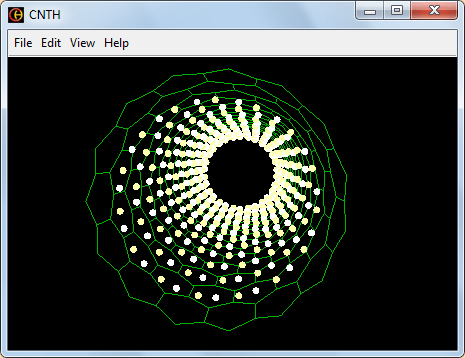
Рис. 5. Вид модели -
Для вызова диалога сохранения воспользуйтесь командой Save as… из меню File.
-
Запустите программу «NanoEvolver» — Dissipative kT (версия 4.10), если она еще не запущена.
-
Загрузите из внешнего файла модель структуры углеродного нанотубулярного аккумулятора водорода. Загрузка осуществляется посредством команды Load structure… из меню File.
-
Расположите модель на экране так, чтобы обеспечить удобство ее обзора. Используйте клавиши ←↑↓→ для вращения модели в пространстве; Insert/Delete — для приближения/отдаления; Page Up/Page Down — для перемещения по вертикали; Home/End — для перемещения по горизонтали.
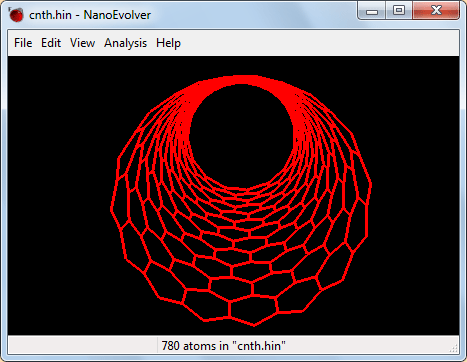
Рис. 6. Вид модели углеродного нанотубулярного аккумулятора водорода в окне «NanoEvolver» -
Из меню View вызовите команду Graph…
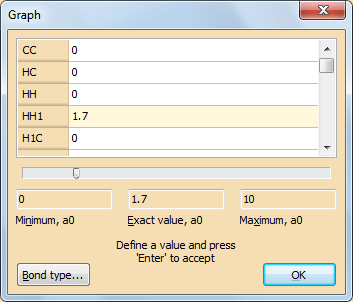
Рис. 7. Диалоговое окно Graph…Затем сохраните структуру с типом файла, как показано на на рис. 8:
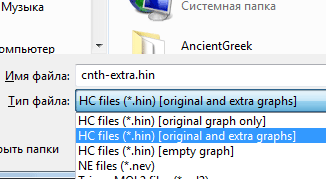
Рис. 8. Выбор формата сохранения структуры -
Откройте сохраненную структуру в программе «NanoEvolver».
-
В «NanoEvolver» используются аппроксимирующие парные потенциалы в форме функции Морзе. Параметры потенциалов — D0 (энергия диссоциации), R0 (равновесное расстояние) и ω (частота колебаний) — чаще всего рассчитываются неэмпирическими методами ab initio, а подходящая аналитическая форма электронным термам придается с использованием метода аппроксимации по дискретным расчетным точкам. В данном случае расчеты параметров всех используемых потенциалов связей в системе выполнены методом нелокального функционала плотности в пакете программ «Winbond».
Задание параметров межатомных потенциалов для загруженной модели и определение установок расчетной процедуры.
Указание параметров потенциалов, а также некоторых важных опций и параметров, влияющих на ход расчетной процедуры, производится в диалоговом окне Settings (см. рис. 9), вызвать которое можно с помощью команды Set parameters… меню Edit.
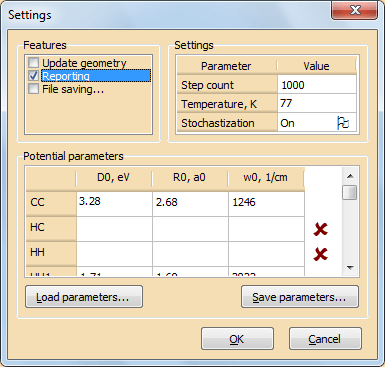
Рис. 9. Диалоговое окно выставления опций и параметровУстановите параметры, как показано на рис. 9. Параметры межатомных потенциалов введите нажав на кнопку Load parameters…, загрузите эти данные из внешнего файла параметров Potential_parameters.txt, который расположен в той же папке, что и загруженная структура.
-
Сохраните отчет во внешнем файле. Для этого вызовите команду Save report as… из меню File. Укажите путь и имя файла, нажмите Сохранить. «NanoEvolver» сохраняет отчеты в удобном формате гипертекста, можете сейчас просмотреть сохраненный на диске отчет, используя любой установленный на компьютере браузер. Анализируя отчет, обратите внимание на единицы измерения и порядок величин.
-
Рис. 10. Окно с кратким отчетом при завершении расчетной процедурыПрежде чем проводить непосредственно сам процесс самоорганизации структуры, в диалоговом окне Settings (см. рис. 9) активируйте опцию File saving. В появившемся окне укажите имя файла, в котором будут сохранены данные необходимые для выявления кинетических закономерностей. Далее раскройте меню Edit главного окна программы «NanoEvolver» и выберите пункт Evolve. В зависимости от количества итераций, выставленного в окне Settings, а также от размеров выбранного для расчетов образца, процедура моделирования процесса самоорганизации может занять несколько минут. По завершении первого «прогона» процедуры отметьте (запомните или запишите) текущие значения полной энергии связи и градиента энергии. В окне Settings установите новое число итераций (например, 500) и прогоните дополнительный расчет. Если в результате повторных расчетов значения энергии и градиента меняются мало, можно переходить к следующему шагу.
-
Оцените визуально изменения, которые произошли в геометрии структуры. Для получения наиболее отчетливой картины деформаций поверхности вы можете вращать и перемещать модель в пространстве, а также подобрать другие цвета для атомов структуры и для фона. Подбор цвета атомов и фона осуществляется посредством команд Define atoms color… и Define window color… соответственно.
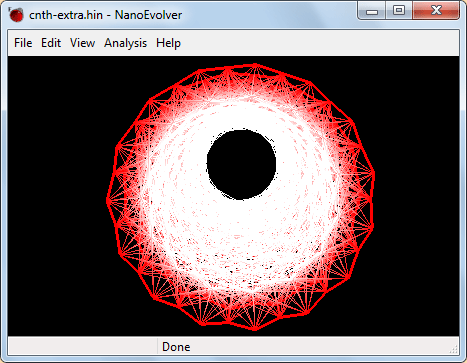
Рис. 11. Геометрия исследуемой системы как результат расчетов в программе «NanoEvolver» -
Сохраните отчет (File→Save report as…) для полученной в результате структуры. Исследуйте отчет. Обратите внимание на изменение полной энергии связи, если таковое имеется, пересчитайте характеристики в единицы СИ.
-
Сохраните на диск модель конечной структуры как результат своих расчетов. Вызовите для этого команду Save structure… из меню File. В диалоговом окне сохранения укажите тип файлов NE files (*.nev) либо HC files (*.hin).
-
При необходимости можно сохранить для отчета графическое изображение модели, как она видна в окне программы «NanoEvolver». Команда сохранения Save image as… находится в пункте File главного меню программы.
-
Откройте файл, в котором сохранены данные необходимые для выявления кинетических закономерностей. Скопируйте все содержимое файла и вставьте в Excel.
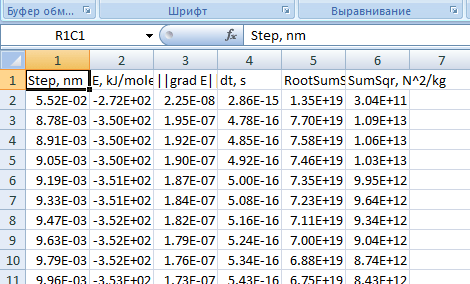
Рис. 12. log-файл, открытый в табличном процессоре Microsoft Office Excel -
Оставьте только те столбцы (см. рис. 13), которые понадобятся для дальнейшей работы.
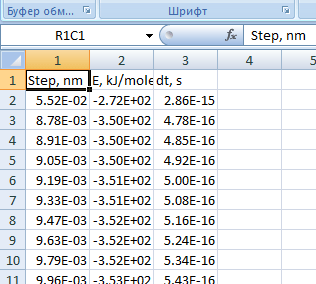
Рис. 13. Оставлены только значимые столбцы отчета -
В следующем пустом столбце необходимо рассчитать интегральное время. Для этого вводите в ячейки данные, как показано на рис. 14.
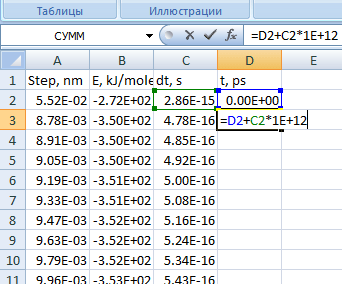
Рис. 14. Окно программы Excel с формулой для счета интегрального времени -
Распространите данную формулу на все стальные ячейки данного столбца.
-
Для удобства проведения дальнейшей работы переведите величину измерения кванта времени из с в фс, используя формулу (см. рис. 15)
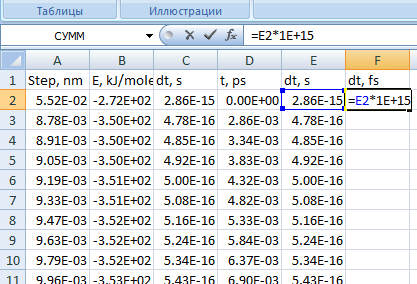
Рис. 15. Окно программы Excel с формулой для пересчета размерности кванта времени -
Постройте развертки эволюции исследуемой системы в координатах
- энергия (E, кДж/моль) — время эволюции (t, пс);
- квант времени (Δt, фс) — время эволюции (t, пс).
Примечание. Указанные пáры вида «A — B» означают, что при построении разверток первая из величин (A) должна быть зависимой переменной и откладываться по оси ординат, а вторая (B) — независимым аргументом, который следует откладывать по оси абсцисс.
-
Проанализируйте вид полученных разверток. Повторите построения для случая температуры, близкой к комнатной (например, T = 293 K). Сделайте выводы. Проведите аналогичное исследование для аккумулятора с другой массовой концентрацией водорода. Сделайте выводы.
III. Литература
- Нечаев Ю.С. О природе, кинетике и предельных значениях сорбции водорода углеродными наноструктурами. УФН 2006, Т. 176. № 6. с. 581–610.
- Nechaev Yu.C. On some techniques and experimental results: relevance for nanotechnology applications. Carbon Nanostructures 2007; 55(11): 189–202.
- Beznosyuk S.A., Vazhenin S.V., Maslova O.A., Zhukovsky M.S., Zhukovsky T.M. Transformation Evolution of Graphene and Nickel Nanoparticles. Carbon Nanomaterials in Clean Energy Hydrogen Systems. NATO Science for Peace and Security Series C: Environmental Security 2008: 215–224.
- Beznosyuk S.A., Maslova O.A., Stobbe I.A., Zhukovsky M.S. Theoretical Modeling of Hydrogen Polycondensation on Carbon Nanotubular Surfaces. J. Nanosci. Nanotechnol., 2009; 9: 1408–1411.
- Beznosyuk S.A., Maslova O.A., Fomina L.V., Zhukovsky M.S. Self-assembling of hydrogen superadsorbate in single-walled carbon nanotubes. Superlatt. and Microstruct. 2009; 46: 384–386.